
Veolia is a leader in developing and delivering effective cooling water corrosion control chemistries.
Corrosion can be defined as the destruction of a metal by chemical or electrochemical reaction with its environment. In cooling systems, corrosion causes two basic problems. The first and most obvious is the failure of equipment with the resultant cost of replacement and plant downtime. The second is decreased plant efficiency due to loss of heat transfer-the result of heat exchanger fouling caused by the accumulation of corrosion products.
Corrosion occurs at the anode, where metal dissolves. Often, this is separated by a physical distance from the cathode, where a reduction reaction takes place. An electrical potential difference exists between these sites, and current flows through the solution from the anode to the cathode. This is accompanied by the flow of electrons from the anode to the cathode through the metal. Figure 24-1 illustrates this process.
For steel, the typical anodic oxidation reaction is:
| Fe = Fe2+ + 2e¯ |
This reaction is accompanied by the following:
| Fe2+ + 2OH¯ = Fe(OH)2 |
The ferrous hydroxide then combines with oxygen and water to produce ferric hydroxide, Fe(OH)3, which becomes common iron rust when dehydrated to Fe2O3.
The primary cathodic reaction in cooling systems is:
| O2 + H2O + 2e¯ = 2OH¯ |
The production of hydroxide ions creates a localized high pH at the cathode, approximately 1-2 pH units above bulk water pH. Dissolved oxygen reaches the surface by diffusion, as indicated by the wavy lines in Figure 24-1. The oxygen reduction reaction controls the rate of corrosion in cooling systems; the rate of oxygen diffusion is usually the limiting factor.
Another important cathodic reaction is:
| 2H+ + 2e¯ = H2 |
At neutral or higher pH, the concentration of H+ ions is too low for this reaction to contribute significantly to the overall corrosion rate. However, as pH decreases, this reaction becomes more important until, at a pH of about 4, it becomes the predominant cathodic reaction.
The formation of anodic and cathodic sites, necessary to produce corrosion, can occur for any of a number of reasons: impurities in the metal, localized stresses, metal grain size or composition differences, discontinuities on the surface, and differences in the local environment (e.g., temperature, oxygen, or salt concentration). When these local differences are not large and the anodic and cathodic sites can shift from place to place on the metal surface, corrosion is uniform (see Figure 24-2). With uniform corrosion, fouling is usually a more serious problem than equipment failure.
Localized corrosion, which occurs when the anodic sites remain stationary, is a more serious industrial problem. Forms of localized corrosion include pitting, selective leaching (e.g., dezincification), galvanic corrosion, crevice or underdeposit corrosion, intergranular corrosion, stress corrosion cracking, and microbiologically influenced corrosion. Another form of corrosion, which cannot be accurately categorized as either uniform or localized, is erosion corrosion.
Pitting
Pitting (see Figure 24-3) is one of the most destructive forms of corrosion and also one of the most difficult to predict in laboratory tests. Pitting occurs when anodic and cathodic sites become stationary due to large differences in surface conditions. It is generally promoted by low-velocity or stagnant conditions (e.g., shell-side cooling) and by the presence of chloride ions. Once a pit is formed, the solution inside it is isolated from the bulk environment and becomes increasingly corrosive with time. The high corrosion rate in the pit produces an excess of positively charged metal cations, which attract chloride anions. In addition, hydrolysis produces H+ ions. The increase in acidity and concentration within the pit promotes even higher corrosion rates, and the process becomes self-sustaining. Inhibitors can be used to control pitting, but they must be applied correctly.
Selective Leaching
Selective leaching is the corrosion of one element of an alloy. The most common example in cooling systems is dezincification, which is the selective removal of zinc from copper-zinc alloys (see Figure 24-4). The conditions that promote the pitting of steel also promote the pitting of brass, which in cooling systems usually occurs by dezincification. Low pH conditions (<6.0) and high free chlorine residuals (>1.0 ppm) are particularly aggressive in producing dezincification. The dezincification resistance varies with the alloy. For example, 70-30 brass is less resistant than admiralty brass (70-30 brass plus 1% tin), which is less resistant than inhibited Admiralty brass (Admiralty brass plus a small amount of arsenic, antimony, or phosphorus).
Galvanic Corrosion
Galvanic corrosion occurs when two dissimilar metals are in contact in a solution. The contact must be good enough to conduct electricity, and both metals must be exposed to the solution. The driving force for galvanic corrosion is the electric potential difference that develops between two metals. This difference increases as the distance between the metals in the galvanic series increases. Table 24-1 shows a galvanic series for some commercial metals and alloys. When two metals from the series are in contact in solution, the corrosion rate of the more active (anodic) metal increases and the corrosion rate of the more noble (cathodic) metal decreases.
Table 24-1. Galvanic series of metals and alloys
Courtesy of International Nickel Company, Inc.
| CORRODED END (anodic, or least noble) |
| Magnesium Magnesium alloys |
| Zinc |
| Aluminum 2S |
| Cadmium |
| Aluminum 17ST |
| Steel or Iron Cast Iron |
| Chromium-iron (active) |
| Ni-Resist 18-8-Cr-Ni-Fe (active) |
| 18-8-3-Cr-Ni-Mo-Fe (active) |
| Hastelloy C |
| Lead-tin Solders Lead Tin |
| Nickel (active) |
| Inconel (active) |
| Hastelloy A Hastelloy B |
| Brasses Copper Bronzes Copper-nickel alloys Titanium Monel |
| Silver Solder |
| Nickel (passive) Inconel (passive) Chromium-iron (passive) 18-8-Cr-Ni-Fe (passive) 18-8-3-Cr-Ni-Mo-Fe (passive) |
| Silver |
| Graphite |
| PROTECTED END (cathodic, or most noble) |
Galvanic corrosion can be controlled by the use of sacrificial anodes. This is a common method of controlling corrosion in heat exchangers with Admiralty tube bundles and carbon steel tube sheets and channel heads. The anodes are bolted directly to the steel and protect a limited area around the anode. Proper placement of sacrificial anodes is a precise science.
The most serious form of galvanic corrosion occurs in cooling systems that contain both copper and steel alloys. It results when dissolved copper plates onto a steel surface and induces rapid galvanic attack of the steel. The amount of dissolved copper required to produce this effect is very small and the increased corrosion is very difficult to inhibit once it occurs. A copper corrosion inhibitor is needed to prevent copper dissolution.
Crevice Corrosion
Crevice corrosion is intense localized corrosion which occurs within a crevice or any area that is shielded from the bulk environment. Solutions within a crevice are similar to solutions within a pit in that they are highly concentrated and acidic. Because the mechanisms of corrosion in the two processes are virtually identical, conditions that promote pitting also promote crevice corrosion. Alloys that depend on oxide films for protection (e.g., stainless steel and aluminum) are highly susceptible to crevice attack because the films are destroyed by high chloride ion concentrations and low pH. This is also true of protective films induced by anodic inhibitors.
The best way to prevent crevice corrosion is to prevent crevices. From a cooling water standpoint, this requires the prevention of deposits on the metal surface. Deposits may be formed by suspended solids (e.g., silt, silica) or by precipitating species, such as calcium salts.
Intergranular Corrosion
Intergranular corrosion is localized attack that occurs at metal grain boundaries. It is most prevalent in stainless steels which have been improperly heat-treated. In these metals, the grain boundary area is depleted in chromium and therefore is less resistant to corrosion (see Figure 24-5). Intergranular corrosion also occurs in certain high-strength aluminum alloys. In general, it is not of significance in cooling systems.
Stress Corrosion Cracking
Stress corrosion cracking (SCC) is the brittle failure of a metal by cracking under tensile stress in a corrosive environment. Failures tend to be transgranular (see Figure 24-6), although inter-granular failures have been noted. Commonly used cooling system alloys that may crack due to stress include austenitic stainless steels (300 series) and brasses. The susceptibility of stainless steels to SCC increases significantly as the temperature is increased. Most laboratory stainless steel SCC testing is done at about 300°F, because it is very difficult to promote cracking at temperatures below 200°F. For this reason, SCC of stainless steels has not been widely observed in cooling systems.
Chloride is the main contributor to SCC of stainless steels. High chloride concentrations, resulting from high chloride levels in the makeup water and/or high cycles of concentration, will increase susceptibility. Although low water temperatures generally preclude cracking, SCC of stainless steels can occur in cooling systems.
For brasses, the ammonium ion is the main cause of SCC. Very few service failures have been reported where ammonia is not present.
The most likely places for SCC to be initiated are crevices or areas where the flow of water is restricted. This is due to the buildup of corrodent concentrations in these areas. For example, chloride can concentrate from 100 ppm in the bulk water to as high as 10,000 ppm (1%) in a crevice. Deposits are initiating sites because of crevices formed beneath them. The low water velocities in shell-side cooling are also detrimental.
The most effective way to prevent SCC in both stainless steel and brass systems is to keep the system clean and free of deposits. An effective deposit control treatment is imperative. A good corrosion inhibitor is also beneficial. Chromate and phosphate have each been used successfully to prevent the SCC of stainless steel in chloride solutions.
Microbiologically Influenced Corrosion (MIC)
Microorganisms in cooling water form "biofilms" on cooling system surfaces. Biofilms consist of populations of sessile organisms and their hydrated polymeric secretions. Numerous types of organisms may exist in any particular biofilm, ranging from strictly aerobic bacteria at the water interface to anaerobic bacteria such as sulfate-reducing bacteria (SRB) at the oxygen-depleted metal surface.
The presence of a biofilm can contribute to corrosion in three ways: physical deposition, production of corrosive by-products, and depolarization of the corrosion cell caused by chemical reactions.
As discussed previously, deposits can cause accelerated localized corrosion by creating differential aeration cells. This same phenomenon occurs with a biofilm. The nonuniform nature of biofilm formation creates an inherent differential, which is enhanced by the oxygen consumption of organisms in the biofilm.
Many of the by-products of microbial metabolism, including organic acids and hydrogen sulfide, are corrosive. These materials can concentrate in the biofilm, causing accelerated metal attack.
Corrosion tends to be self-limiting due to the buildup of corrosion reaction products. However, microbes can absorb some of these materials in their metabolism, thereby removing them from the anodic or cathodic site. The removal of reaction products, termed "depolarization," stimulates further corrosion.
Figure 24-7 shows a typical result of microbial corrosion. The surface exhibits scattered areas of localized corrosion, unrelated to flow pattern. The corrosion appears to spread in a somewhat circular pattern from the site of initial colonization.
Erosion Corrosion
Erosion corrosion is the increase in the rate of metal deterioration from abrasive effects. It can be identified by grooves and rounded holes, which usually are smooth and have a directional pattern. Erosion corrosion is increased by high water velocities and suspended solids. It is often localized at areas where water changes direction. Cavitation (damage due to the formation and collapse of bubbles in high-velocity turbines, propellers, etc.) is a form of erosion corrosion. Its appearance is similar to closely spaced pits, although the surface is usually rough.
Corrosion control requires a change in either the metal or the environment. The first approach, changing the metal, is expensive. Also, highly alloyed materials, which are very resistant to general corrosion, are more prone to failure by localized corrosion mechanisms such as stress corrosion cracking.
The second approach, changing the environment, is a widely used, practical method of preventing corrosion. In aqueous systems, there are three ways to effect a change in environment to inhibit corrosion:
- form a protective film of calcium carbonate on the metal surface using the natural calcium and alkalinity in the water
- remove the corrosive oxygen from the water, either by mechanical or chemical deaeration
- add corrosion inhibitors
Calcium Carbonate Protective Scale
The Langelier Saturation Index (LSI) is a useful tool for predicting the tendency of a water to deposit or dissolve calcium carbonate (see Chapter 25 for a thorough discussion of LSI). A uniform coating of calcium carbonate, deposited on the metal surfaces, physically segregates the metal from the corrosive environment. To develop the positive LSI required to deposit calcium carbonate, it is usually necessary to adjust the pH, alkalinity, or calcium content of the water. Soda ash, caustic soda, or lime (calcium hydroxide) may be used for this adjustment. Lime is usually the most economical alkali because it raises the calcium content as well as the alkalinity and pH.
Theoretically, controlled deposition of calcium carbonate scale can provide a film thick enough to protect, yet thin enough to allow adequate heat transfer. However, low-temperature areas do not permit the development of sufficient scale for corrosion protection, and excessive scale forms in high-temperature areas and interferes with heat transfer. Therefore, this approach is not used for industrial cooling systems. Controlled calcium carbonate deposition has been used successfully in some waterworks distribution systems where substantial temperature increases are not encountered.
Mechanical and Chemical Deaeration
The corrosive qualities of water can be reduced by deaeration. Vacuum deaeration has been used successfully in once-through cooling systems. Where all oxygen is not removed, catalyzed sodium sulfite can be used to remove the remaining oxygen. The sulfite reaction with dissolved oxygen is:
Na2SO3 + 1/2 O2 = Na2SO4
sodium oxygen sodium
sulfite sulfate
The use of catalyzed sodium sulfite for chemical deaeration requires 8 parts of catalyzed sodium sulfite for each part of dissolved oxygen. In certain systems where vacuum deaeration is already used, the application of catalyzed sodium sulfite may be economically justified for removal of the remaining oxygen. The use of sodium sulfite may also be applicable to some closed loop cooling systems.
In open recirculating cooling systems, continual replenishment of oxygen as the water passes over the cooling tower makes deaeration impractical.
Corrosion Inhibitors
A corrosion inhibitor is any substance which effectively decreases the corrosion rate when added to an environment. An inhibitor can be identified most accurately in relation to its function: removal of the corrosive substance, passivation, precipitation, or adsorption.
- Deaeration (mechanical or chemical) removes the corrosive substance-oxygen.
- Passivating (anodic) inhibitors form a pro-tective oxide film on the metal surface. They are the best inhibitors because they can be used in economical concentrations, and their protective films are tenacious and tend to be rapidly repaired if damaged (see Figure 24-8).
- Precipitating (cathodic) inhibitors are simply chemicals which form insoluble precipitates that can coat and protect the surface. Precipitated films are not as tenacious as passive films and take longer to repair after a system upset.
A discussion of each of these inhibitors follows, preceded by an overview of the role of polarization in corrosion.
Polarization. Figure 24-9 is a schematic diagram showing common potential vs. corrosion current plots. The log current is the rate of electrochemical reaction, and the plots show how the rate of anodic and cathodic reactions change as a function of surface potential. In Figure 24-9(a), uninhibited corrosion is occurring. The corrosion potential, Ecorr, and the corrosion current, Icorr, are defined by the point at which the rate of the anodic reaction equals the rate of the cathodic reaction. Icorr is the actual rate of metal dissolution. Figure 24-9(b) shows the condition after an anodic inhibitor has been applied. The rate of the anodic reaction has been decreased. This causes a decrease in Icorr accompanied by a shift in Ecorr to a more positive (anodic) potential. Figure 24-9(c) shows the effect of a cathodic inhibitor. Here, the rate of the cathodic reaction has been decreased, accompanied again by a decrease in Icorr, but this time the shift in Ecorr is in the negative (cathodic) direction.
Passivation Inhibitors. Examples of passivators (anodic inhibitors) include chromate, nitrite, molybdate, and orthophosphate. All are oxidizers and promote passivation by increasing the electrical potential of the iron. Chromate and nitrite do not require oxygen and thus can be the most effective. Chromate is an excellent aqueous corrosion inhibitor, particularly from a cost perspective. However, due to health and environmental con-cerns, use of chromate has decreased significantly and will probably be outlawed in the near future. Nitrite is also an effective inhibitor, but in open systems it tends to be oxidized to nitrate.
Both molybdate and orthophosphate are excellent passivators in the presence of oxygen. Molybdate can be a very effective inhibitor, especially when combined with other chemicals. Its main drawback is its high cost. Orthophosphate is not really an oxidizer per se, but becomes one in the presence of oxygen. If iron is put into a phosphate solution without oxygen present, the corrosion potential remains active and the corrosion rate is not reduced. However, if oxygen is present, the corrosion potential increases in the noble direction and the corrosion rate decreases significantly.
A negative attribute of orthophosphate is its tendency to precipitate with calcium hardness found in natural waters. In recent years, deposit control agents that prevent this deposition have been developed. Due to its relatively low cost, orthophosphate is widely used as an industrial corrosion inhibitor.
Precipitating Inhibitors. As discussed earlier, the localized pH at the cathode of the corrosion cell is elevated due to the generation of hydroxide ions. Precipitating inhibitors form complexes which are insoluble at this high pH (1-2 pH units above bulk water), but whose deposition can be controlled at the bulk water pH (typically 7-9 pH). A good example is zinc, which can precipitate as hydroxide, carbonate, or phosphate. Calcium carbonate and calcium orthophosphate are also precipitating inhibitors. Orthophosphate thus exhibits a dual mechanism, acting as both an anodic passivator and a cathodic precipitator.
Copper Corrosion Inhibitors. The most effective corrosion inhibitors for copper and its alloys are the aromatic triazoles, such as benzotriazole (BZT) and tolyltriazole (TTA). These compounds bond directly with cuprous oxide (Cu2O) at the metal surface, forming a "chemisorbed" film. The plane of the triazole lies parallel to the metal surface; thus, each molecule covers a relatively large surface area. The exact mechanism of inhibition is unknown. Various studies indicate anodic inhibition, cathodic inhibition, or a combination of the two. Other studies indicate the formation of an insulating layer between the water surface and the metal surface. A very recent study supports the idea of an electronic stabilization mechanism. The protective cuprous oxide layer is prevented from oxidizing to the nonprotective cupric oxide. This is an anodic mechanism. However, the triazole film exhibits some cathodic properties as well.
In addition to bonding with the metal surface, triazoles bond with copper ions in solution. Thus, dissolved copper represents a "demand" for triazole, which must be satisfied before surface filming can occur. Although the surface demand for triazole filming is generally negligible, copper corrosion products can consume a considerable amount of treatment chemical. Excessive chlorination will deactivate the triazoles and significantly increase copper corrosion rates. Due to all of these factors, treatment with triazoles is a complex process.
Adsorption Inhibitors. Adsorption inhibitors must have polar properties in order to be adsorbed and block the surface against further adsorption. Typically, they are organic compounds containing nitrogen groups, such as amines, and organic compounds containing sulfur or hydroxyl groups. The size, orientation, shape, and electrical charge distribution of the molecules are all important factors. Often, these molecules are surfactants and have dual functionality. They contain a hydrophilic group, which adsorbs onto the metal surface, and an opposing hydrophobic group, which prevents further wetting of the metal.
Glycine derivatives and aliphatic sulfonates are examples of compounds which can function in this way. The use of these inhibitors in cooling systems is usually limited by their biodegradability and their toxicity toward fish. In addition, they can form thick, oily surface films, which may severely retard heat transfer.
Silicates. For many years, silicates have been used to inhibit aqueous corrosion, particularly in potable water systems. Probably due to the complexity of silicate chemistry, their mechanism of inhibition has not yet been firmly established. They are nonoxidizing and require oxygen to inhibit corrosion, so they are not passivators in the classical sense. Yet they do not form visible precipitates on the metal surface. They appear to inhibit by an adsorption mechanism. It is thought that silica and iron corrosion products interact. However, recent work indicates that this interaction may not be necessary. Silicates are slow-acting inhibitors; in some cases, 2 or 3 weeks may be required to establish protection fully. It is believed that the polysilicate ions or colloidal silica are the active species and these are formed very slowly from monosilicic acid, which is the predominant species in water at the pH levels maintained in cooling systems.
Effect of Conductivity, pH, and Dissolved Oxygen
Figures 24-10 through 24-12 show the effects of several operating parameters on the corrosion tendency in aqueous systems. As shown in Figure 24-10, corrosion rate increases with conductivity.
Figure 24-11 shows the effect of pH on the corrosion of iron. Within the acid range (pH <4), the iron oxide film is continually dissolved. In cooling water, the potential for calcium carbonate precipitation increases with higher pH and alkalinity; thus the corrosion rate decreases slightly as pH is increased from 4 to 10. Above pH 10, iron becomes increasingly passive.
Figure 24-12 shows the effect of oxygen concentration on corrosion at different temperatures. As discussed previously, oxygen is the main driving force for corrosion of steel in cooling water. The increase in corrosion with temperature at a given oxygen concentration is due to more rapid oxygen diffusion occurring at higher temperatures.
Practical Considerations
The success of cooling water corrosion inhibitor programs is affected by the following factors:
- Water Characteristics. Calcium, alkalinity, and pH levels in water are important factors for reasons already cited. Further discussions are covered in the chapters on once-through and open recirculating systems.
- Design Considerations. Low water velocity, which occurs in shell-side cooling, increases deposition. This factor must be addressed in the design of the system.
- Microbiological Control. An effective microbiological control program is necessary to prevent severe fouling problems. Fouling caused by uncontrolled biological growth can contribute to corrosion by one or more mechanisms.
- System Control. Even the best treatment technology available will fail without a reasonable level of control. Therefore, careful consideration must be given to system control-the accuracy with which the pH, inhibitor levels, and other water character-istics are maintained.
- Pretreatment. Grease and/or corrosion products from previous treatment programs should be cleaned out, and the system should be treated with a high level of a good inhibitor before normal operation.
- Contamination. Contamination can also be a problem. Sulfide, ammonia, and hydrocarbons are among the most severe contaminants. Sulfide is corrosive to steel and copper alloys. Ammonia is corrosive to Admiralty and promotes biological growth. Hydrocarbons promote fouling and biological growth.
In the determination of treatment levels, solubility data is important. The Langelier Saturation Index, which defines the solubility of calcium carbonate, is commonly used. Solubility data for calcium orthophosphate and zinc orthophosphate may be needed if the treatment contains phosphate and zinc.
Monitoring
Every cooling water system should include a method of monitoring corrosion in the system. Tools commonly used for this purpose include metal corrosion coupons, instantaneous corrosion rate meters, and heated surfaces such as test heat exchangers and the Betz MonitAll® apparatus. Data obtained from these devices can be used to optimize an inhibitor treatment program to maintain the plant equipment in the best possible condition. When heat transfer data cannot be obtained on operating exchangers, monitoring devices can be useful for evaluating the success of a treatment program without a plant shutdown.
Corrosion Coupons. Preweighed metal coupons are still widely used as a reliable method for monitoring corrosion in cooling systems. Coupon weight loss provides a quantitative measure of the corrosion rate, and the visual appearance of the coupon provides an assessment of the type of corrosion and the amount of deposition in the system. In addition, measurement of pit depths on the coupon can indicate the severity of the pitting.
Coupons should be installed properly in a corrosion coupon bypass rack with continuous, controlled water flow past the coupons. The metallurgy should match that of the system. One disadvantage of coupons is their lack of heat transfer, resulting in a lower temperature than that of the actual heat exchanger tubes. In addition, only a time-weighted average corrosion rate is obtained.
Corrosion Rate Meters. Additional corrosion monitoring tools have been developed by various instrument manufacturers and water treatment companies. Instantaneous corrosion rate meters can measure the corrosion rate at any given point in time.
Instrument methods fall into two general categories: electrical resistance and linear polarization. With either technique, corrosion measurements are made quickly without removal of the sensing device.
The electrical resistance method is based on measuring the increase in the electrical resistance of a test electrode as it becomes thinner due to corrosion. This method is desirable because the probes can be installed in both aqueous and nonaqueous streams. However, the electrical resistance method also has its disadvantages: conductive deposits forming on the probe can create misleading results, temperature fluctuations must be compensated for, and pitting character-istics cannot be determined accurately.
The method based on linear polarization at low applied potentials provides instantaneous corrosion rate data that can be read directly from the instrument face in actual corrosion rate units (mils per year). Systems using two or three electrodes are available. This method offers the maximum in performance, simplicity, and reliability.
Corrosion rate meters can be used to assess changes in the corrosion rate as a function of time. They are able to respond to sudden changes in system conditions, such as acid spills, chlorine levels, and inhibitor treatment levels. Coupled with recording devices, they can be powerful tools in diagnosing the causes of corrosion or optimizing inhibitor treatment programs (see Figure 24-13).
Test Heat Exchangers. Test heat exchangers are small exchangers that can be set up to simulate operating conditions in the plant. They provide a convenient way to evaluate corrosion and fouling tendencies on heat transfer surfaces and to measure changes in heat transfer efficiency. A typical design uses cooling water on the tube side and condensing steam as a heat source on the shell side. If the test heat exchanger is insulated, a meaningful "U" (overall heat transfer coefficient) can be calculated.
BETZ MonitAll® Apparatus. The BETZ MonitAll (see Figure 24-14) is designed to measure corrosion and deposition under heat transfer conditions. Cooling water flows over a heated tube section within a glass shell. The specimen tube section is slid onto an electrical heater probe. Thermocouples measure bulk water temperature and tubeside "skin" temperature. The heat flux and flow velocity can be varied to simulate plant conditions. The tubes are available in various metallurgies and are preweighed for corrosion rate determination. The tube is visible through the glass enclosure, allowing direct observation of corrosion and scaling tendencies. Scaling/fouling can be quantified through temperature and flow measurements.
Cooling System Monitoring Station. The Cosmos is a portable data acquisition station that monitors key parameters of a cooling system. The piping and instrumentation cabinet includes flow, pH, and conductivity sensors as well as a corrosion coupon rack, a corrosion rate probe, and a MonitAll unit. Data from all of these devices is fed into the data acquisition system (see Figure 24-15). The accumulated data can be printed directly by the built-in printer or can be downloaded to a personal computer for spreadsheet analysis.
Learn more about Veolia's corrosion control programs.
Figure 24-1. Classic corrosion cell.
Figure 24-2. Uniform corrosion of a mild steel coupon.

Figure 24-3. Pitting can lead to rapid equipment failure.

Figure 24-4. Dezincification of Admiralty brass.

Figure 24-5. Photomicrograph illustrating intergranular corrosion of stainless steel.
Figure 24-6. A transgranular stress corrosion cracking failure.
Figure 24-7. A classic case of biologically induced corrosion.

Figure 24-8. Mild steel corrosion protection provided by a passivationg inhibitor.
Figure 24-9. Simplified polarization curves showing potential vs. log current.
Figure 24-10. As conductivity increases, so does the corrosion rate.
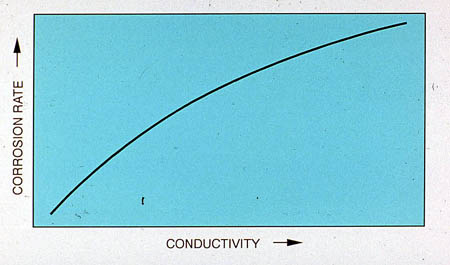
Figure 24-11. The effect of pH on mild steel corrosion rate in an open recirculating cooling system.
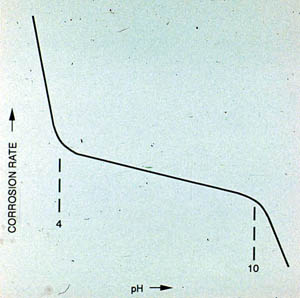
Figure 24-12. Effect of oxygen concentration on corrosion at different temperatures.
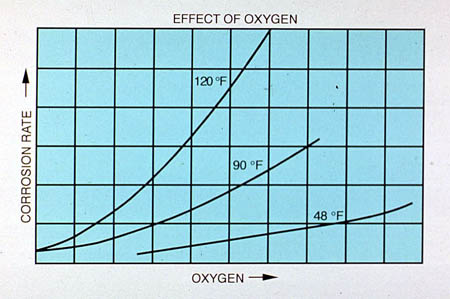
Figure 24-13. Portable corrosion rate meter with recorder.

Figure 24-14. Betz Monitall®

Figure 24-15. Betz COSMOSTMdata acquisition cabinet.
