
System scaling and deposition can cause many problems within your operation. Learn more about Veolia's deposition and scale control chemicals.
Deposit accumulations in cooling water systems reduce the efficiency of heat transfer and the carrying capacity of the water distribution system. In addition, the deposits cause oxygen differential cells to form. These cells accelerate corrosion and lead to process equipment failure. Deposits range from thin, tightly adherent films to thick, gelatinous masses, depending on the depositing species and the mechanism responsible for deposition.
Deposit formation is influenced strongly by system parameters, such as water and skin temperatures, water velocity, residence time, and system metallurgy. The most severe deposition is encountered in process equipment operating with high surface temperatures and/or low water velocities. With the introduction of high-efficiency film fill, deposit accumulation in the cooling tower packing has become an area of concern (see Figure 25-1). Deposits are broadly categorized as scale or foulants.
Scale deposits are formed by precipitation and crystal growth at a surface in contact with water. Precipitation occurs when solubilities are exceeded either in the bulk water or at the surface. The most common scale-forming salts that deposit on heat transfer surfaces are those that exhibit retrograde solubility with temperature.
Although they may be completely soluble in the lower-temperature bulk water, these compounds (e.g., calcium carbonate, calcium phosphate, and magnesium silicate) supersaturate in the higher-temperature water adjacent to the heat transfer surface and precipitate on the surface.
Scaling is not always related to temperature. Calcium carbonate and calcium sulfate scaling occur on unheated surfaces when their solubilities are exceeded in the bulk water (see Figure 25-2). Metallic surfaces are ideal sites for crystal nucleation because of their rough surfaces and the low velocities adjacent to the surface. Corrosion cells on the metal surface produce areas of high pH, which promote the precipitation of many cooling water salts. Once formed, scale deposits initiate additional nucleation, and crystal growth proceeds at an accelerated rate.
Scale control can be achieved through operation of the cooling system at subsaturated conditions or through the use of chemical additives.
Operational Control
The most direct method of inhibiting formation of scale deposits is operation at subsaturation conditions, where scale-forming salts are soluble. For some salts, it is sufficient to operate at low cycles of concentration and/or control pH. However, in most cases, high blowdown rates and low pH are required so that solubilities are not exceeded at the heat transfer surface. In addition, it is necessary to maintain precise control of pH and concentration cycles. Minor variations in water chemistry or heat load can result in scaling (see Figure 25-3).
Chemical Additives
Scaling can be controlled effectively by the use of sequestering agents and chelates, which are capable of forming soluble complexes with metal ions. The precipitation properties of these complexes are not the same as those of the metal ions. Classic examples of these materials are ethylenediaminetetraacetic acid (EDTA) for chelating calcium hardness, and polyphosphates for iron (Figure 25-4). This approach requires stoichiometric chemical quantities. Therefore, its use is limited to waters containing low concentrations of the metal.
Threshold Inhibitors. Deposit control agents that inhibit precipitation at dosages far below the stoichiometric level required for sequestration or chelation are called "threshold inhibitors." These materials affect the kinetics of the nucleation and crystal growth of scale-forming salts, and permit supersaturation without scale formation.
Threshold inhibitors function by an adsorption mechanism. As ion clusters in solution become oriented, metastable microcrystallites (highly oriented ion clusters) are formed. At the initial stage of precipitation, the microcrystallite can either continue to grow (forming a larger crystal with a well defined lattice) or dissolve. Threshold inhibitors prevent precipitation by adsorbing on the newly emerging crystal, blocking active growth sites. This inhibits further growth and favors the dissolution reaction. The precipitate dissolves and releases the inhibitor, which is then free to repeat the process.
Threshold inhibitors delay or retard the rate of precipitation. Crystals eventually form, depending on the degree of supersaturation and system retention time. After stable crystals appear, their continued growth is retarded by adsorption of inhibitor. The inhibitor blocks much of the crystal surface, causing distortions in the crystal lattice as growth continues. The distortions (defects in the crystal lattice) create internal stresses, making the crystal fragile. Tightly adherent scale deposits do not form, because crystals that form on surfaces in contact with flowing water cannot withstand the mechanical force exerted by the water. The adsorbed inhibitor also disperses particles, by virtue of its electrostatic charge, and prevents the formation of strongly bound agglomerates.
The most commonly used scale inhibitors are low molecular weight acrylate polymers and organophosphorus compounds (phosphonates). Both classes of materials function as threshold inhibitors; however, the polymeric materials are more effective dispersants. Selection of a scale control agent depends on the precipitating species and its degree of supersaturation. The most effective scale control programs use both a precipitation inhibitor and a dispersant. In some cases this can be achieved with a single component (e.g., polymers used to inhibit calcium phosphate at near neutral pH).
Langelier Saturation Index
Work by Professor W.F. Langelier, published in 1936, deals with the conditions at which a water is in equilibrium with calcium carbonate. An equation developed by Langelier makes it possible to predict the tendency of calcium carbonate either to precipitate or to dissolve under varying conditions. The equation expresses the relationship of pH, calcium, total alkalinity, dissolved solids, and temperature as they relate to the solubility of calcium carbonate in waters with a pH of 6.5-9.5:
| pHs = (pK2 - pKs) + pCa2+ + pAlk |
where:
pHs = the pH at which water with a given calcium content and alkalinity is in equilibrium with calcium carbonate
K2 = the second dissociation constant for carbonic acid
Ks = the solubility product constant for calcium carbonate
These terms are functions of temperature and total mineral content. Their values for any given condition can be computed from known thermodynamic constants. Both the calcium ion and the alkalinity terms are the negative logarithms of their respective concentrations. The calcium content is molar, while the alkalinity is an equivalent concentration (i.e., the titratable equivalent of base per liter). The calculation of the pHs has been simplified by the preparation of various nomographs. A typical one is shown in Figure 25-5.
The difference between the actual pH (pHa) of a sample of water and the pHs , or pHa - pHs, is called the Langelier Saturation Index (LSI). This index is a qualitative indication of the tendency of calcium carbonate to deposit or dissolve. If the LSI is positive, calcium carbonate tends to deposit. If it is negative, calcium carbonate tends to dissolve. If it is zero, the water is at equilibrium.
The LSI measures only the directional tendency or driving force for calcium carbonate to precipitate or dissolve. It cannot be used as a quantitative measure. Two different waters, one of low hardness (corrosive) and the other of high hardness (scale-forming), can have the same Saturation Index.
The Stability Index developed by Ryzner makes it possible to distinguish between two such waters. This index is based on a study of actual operating results with waters having various Saturation Indexes.
Stability Index = 2(pHs) - pH a
Where waters have a Stability Index of 6.0 or less, scaling increases and the tendency to corrode decreases. Where the Stability Index exceeds 7.0, scaling may not occur at all. As the Stability Index rises above 7.5 or 8.0, the probability of corrosion increases. Use of the LSI together with the Stability Index contributes to more accurate prediction of the scaling or corrosive tendencies of a water.
Fouling occurs when insoluble particulates suspended in recirculating water form deposits on a surface. Fouling mechanisms are dominated by particle-particle interactions that lead to the formation of agglomerates.
At low water velocities, particle settling occurs under the influence of gravity (see Figure 25-6). Parameters that affect the rate of settling are particle size, relative liquid and particle densities, and liquid viscosity. The relationships of these variables are expressed by Stokes' Law. The most important factor affecting the settling rate is the size of the particle. Because of this, the control of fouling by preventing agglomeration is one of the most fundamental aspects of deposition control.
Foulants enter a cooling system with makeup water, airborne contamination, process leaks, and corrosion. Most potential foulants enter with makeup water as particulate matter, such as clay, silt, and iron oxides (see Figure 25-7). Insoluble aluminum and iron hydroxides enter a system from makeup water pretreatment operations. Some well waters contain high levels of soluble ferrous iron that is later oxidized to ferric iron by dissolved oxygen in the recirculating cooling water. Because it is very insoluble, the ferric iron precipitates. The steel corrosion process is also a source of ferrous iron and, consequently, contributes to fouling.
Both iron and aluminum are particularly troublesome because of their ability to act as coagulants. Also, their soluble and insoluble hydroxide forms can each cause precipitation of some water treatment chemicals, such as orthophosphate.
Airborne contaminants usually consist of clay and dirt particles but can include gases such as hydrogen sulfide, which forms insoluble precipitates with many metal ions. Process leaks introduce a variety of contaminants that accelerate deposition and corrosion.
Foulants, such as river water silt, enter the system as finely dispersed particles, which can be as small as 1-100 nm. The particles carry an electrostatic charge, which causes similarly charged particles to repel each other, favoring their dispersion. The net charge a particle carries depends on the composition of the water. Cycling of cooling water increases the concentration of counter-charged ions capable of being electrostatically attracted to and adsorbed onto a charged particle. As counterions adsorb, the net charge of the particle decreases. Particles begin to agglomerate and grow in size as their repulsive forces are diminished.
Settling occurs when the energy imparted by fluid velocity can no longer suspend the particle, due to agglomeration and growth. After particles have settled, the nature of the deposit depends on the strength of the attractive forces between the particles themselves (agglomerate strength) and between the particles and the surface they contact. If attractive forces between particles are strong and the particles are not highly hydrated, deposits are dense and well structured; if the forces are weak, the deposits are soft and pliable. Deposition continues as long as the shear strength of the deposit exceeds the shear stress of the flowing water.
Methods of fouling control are discussed in the following sections.
Removal of Particulate Matter
The amount of particulate entering a cooling system with the makeup water can be reduced by filtration and/or sedimentation processes. Particulate removal can also be accomplished by filtration of recirculating cooling water. These methods do not remove all of the suspended matter from the cooling water. The level of fouling experienced is influenced by the effectiveness of the particular removal scheme employed, the water velocities in the process equipment, and the cycles of concentration maintained in the cooling tower.
High Water Velocities
The ability of high water velocities to minimize fouling depends on the nature of the foulant. Clay and silt deposits are more effectively removed by high water velocities than aluminum and iron deposits, which are more tacky and form interlocking networks with other precipitates. Operation at high water velocities is not always a viable solution to clay and silt deposition because of design limitations, economic considerations, and the potential for erosion corrosion.
Dispersants
Dispersants are materials that suspend particulate matter by adsorbing onto the surface of particles and imparting a high charge. Electrostatic repulsion between like-charged particles prevents agglomeration, which reduces particle growth. The presence of a dispersant at the surface of a particle also inhibits the bridging of particles by precipitates that form in the bulk water. The adsorption of the dispersant makes particles more hydrophilic and less likely to adhere to surfaces. Thus, dispersants affect both particle-to-particle and particle-to-surface interactions.
The most effective and widely used dispersants are low molecular weight anionic polymers. Dispersion technology has advanced to the point at which polymers are designed for specific classes of foulants or for a broad spectrum of materials. Acrylate-based polymers are widely used as dispersants. They have advanced from simple homopolymers of acrylic acid to more advanced copolymers and terpolymers. The performance characteristics of the acrylate polymers are a function of their molecular weight and structure, along with the types of monomeric units incorporated into the polymer backbone.
Surfactants
Surface-active or wetting agents are used to prevent fouling by insoluble hydrocarbons. They function by emulsifying the hydrocarbon through the formation of microdroplets containing the surfactant. The hydrophobic (water-hating) portion of the surfactant is dissolved within the oil drop, while the hydrophilic (water-loving) portion is at the surface of the droplet. The electrostatic charge imparted by hydrophilic groups causes the droplets to repel each other, preventing coalescence.
Through a similar process, surfactants also assist in the removal of hydrocarbon-containing deposits.
Learn more about Veolia's deposition and scale control programs.
Figure 25-1. Scaling of cooling tower fill by a combination of calcium carbonate and calcium phosphate.

Figure 25-2. Scaling of water distribution piping by calcium sulfate.

Figure 25-3. Calcium carbonate scaling of an electric utility surface condenser due to poor pH control.
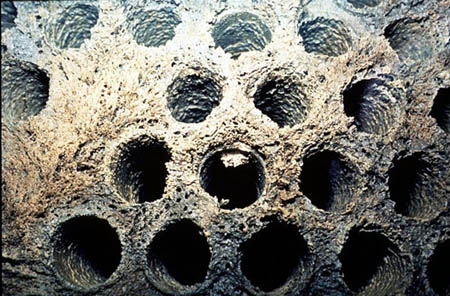
Figure 25-4. Sequestration of iron by sodium phosphates.
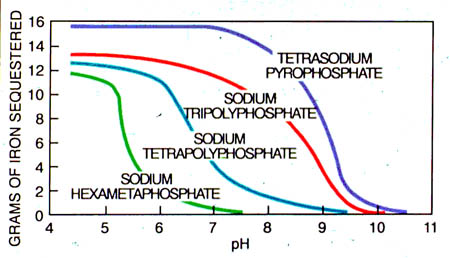
Figure 25-5. Langelier Saturation Index Chart
Figure 25-6. Calcium and iron phosphate fouling due to low water velocity.

Figure 25-7. Iron and silt fouling of plate cooler.
